四大计算方向,
专业计算团队支持。
点击卡片展开详情,了解每个方向能帮你做什么。
量子化学计算
:孤立分子、离子、团簇(≤400 原子),不涉及周期性边界条件。
:密度泛函理论(DFT)、后HF方法、半经验方法。
:结构优化与振动分析、过渡态搜索与 IRC 验证、HOMO/LUMO 前线轨道分析、静电势与福井函数反应性预测、激发态 TDDFT 与荧光/磷光光谱。
量子化学通过求解分子体系的薛定谔方程,获得几何构型、电子分布、热力学量及光谱特性。我们从快速预筛的半经验方法到高精度后HF计算,根据研究需要匹配理论级别。默认使用经业内验证的泛函与基组,全部结果配套可发表级 SI 文档并支持审稿回复。
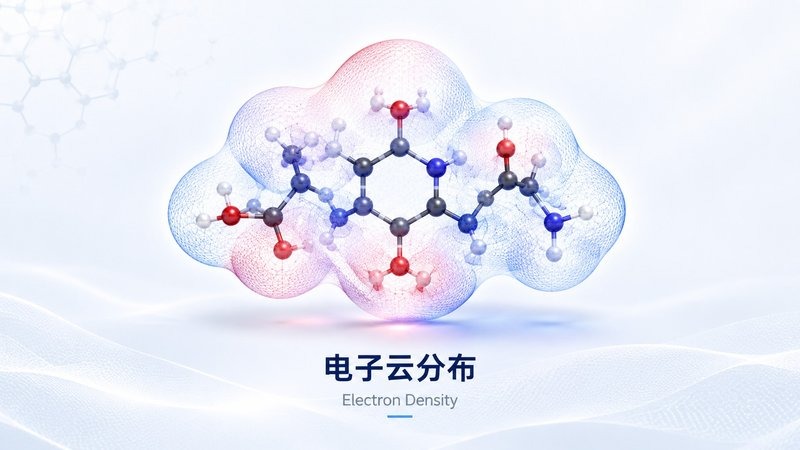 电子云分布
电子云分布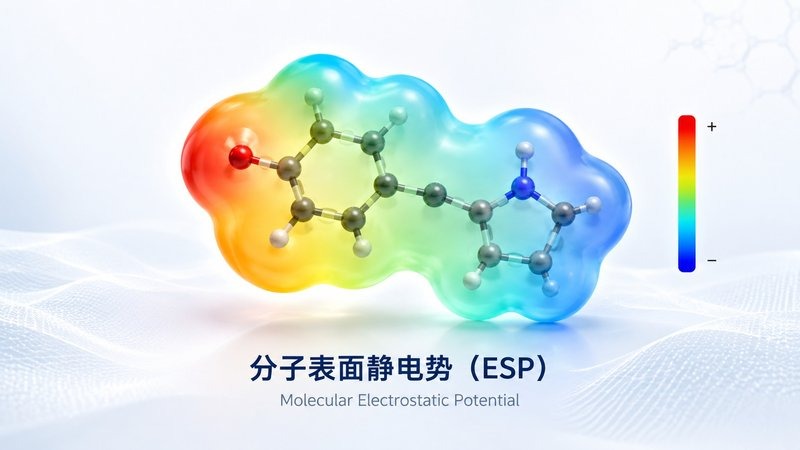 分子表面静电势
分子表面静电势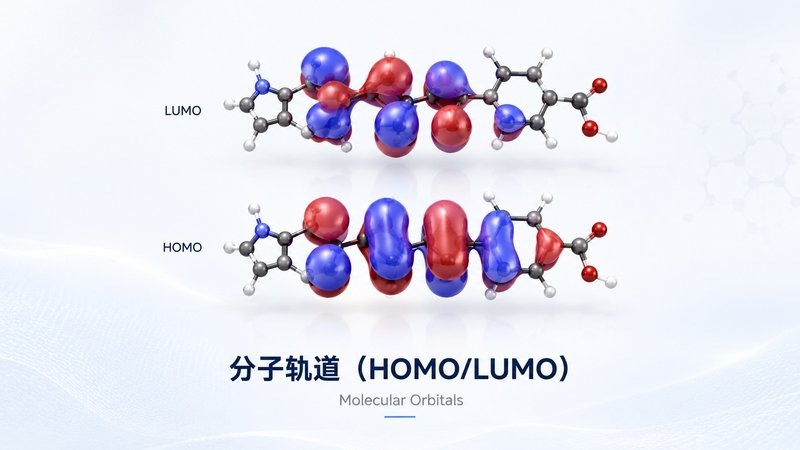 分子轨道 HOMO/LUMO
分子轨道 HOMO/LUMO 振动模态分析
振动模态分析 溶剂化效应 PCM
溶剂化效应 PCM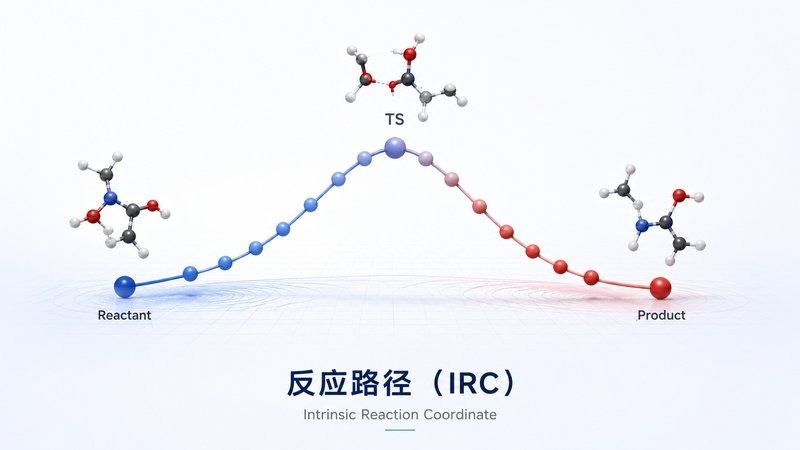 反应路径 IRC
反应路径 IRC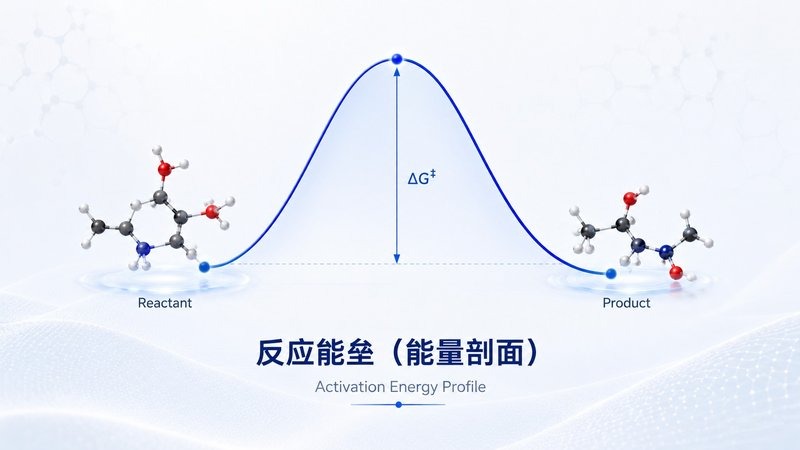 反应能垒
反应能垒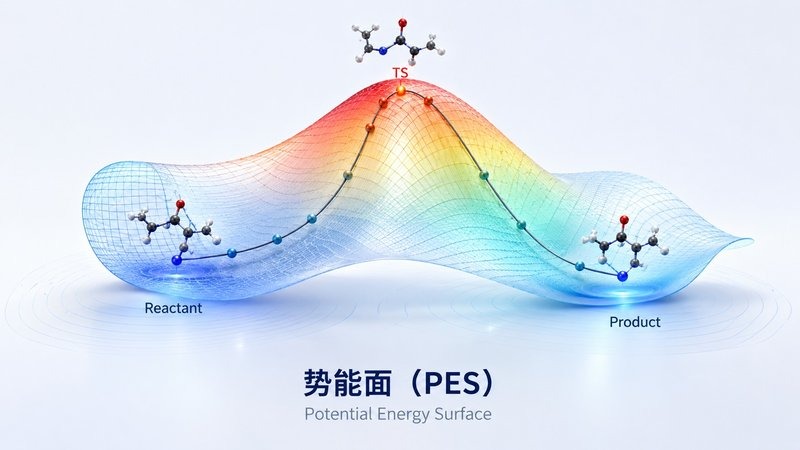 势能面 PES
势能面 PES从小分子到过渡金属配合物,覆盖结构优化、过渡态搜索、IRC 反应路径、振动分析与热力学量、静电势与前线轨道分析。可发表级 SI 文档,支持审稿回复。
- 结构优化 & 振动分析(热力学量)
- 过渡态搜索 & IRC 路径验证
- HOMO/LUMO 前线轨道分析
- 静电势 & 福井函数反应性预测
- 激发态 TDDFT、荧光磷光
第一性原理计算
:晶体、表面、二维材料、异质结等周期性体系(晶胞 ≤400 原子,CP2K 可扩展至 1000 原子),不适用于孤立分子体系。
:周期性密度泛函理论(DFT),平面波/高斯基组。
:体相弛豫与电子结构、表面 slab 模型与表面能、能带结构与 PDOS 分析、COHP/COOP 化学键分析、吸附能与功函数、Bader 电荷与电荷密度差。
第一性原理计算无需经验参数,从原子坐标出发预测材料的电子结构与理化性质,适用于催化剂设计、电池材料、半导体器件等场景。k 点收敛测试与截断能优化确保结果可靠,数据与图表可直接用于论文发表。
 吸附构型 — 分子在晶体表面吸附
吸附构型 — 分子在晶体表面吸附 不同吸附位点 — 顶位、桥位、空位、π键合
不同吸附位点 — 顶位、桥位、空位、π键合 表面Slab模型 — 周期性格子与真空层
表面Slab模型 — 周期性格子与真空层 表面重构 — 表面原子自发重排
表面重构 — 表面原子自发重排 迁移路径 — 离子扩散能垒与能量景观
迁移路径 — 离子扩散能垒与能量景观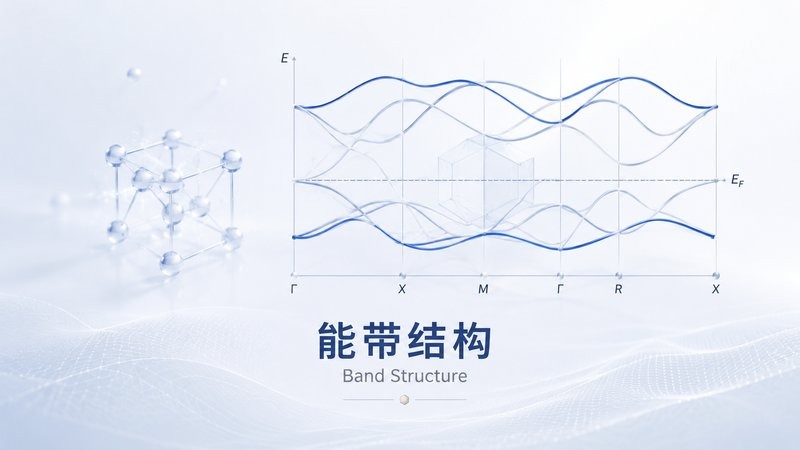 能带结构 — 晶体能带色散与费米能级
能带结构 — 晶体能带色散与费米能级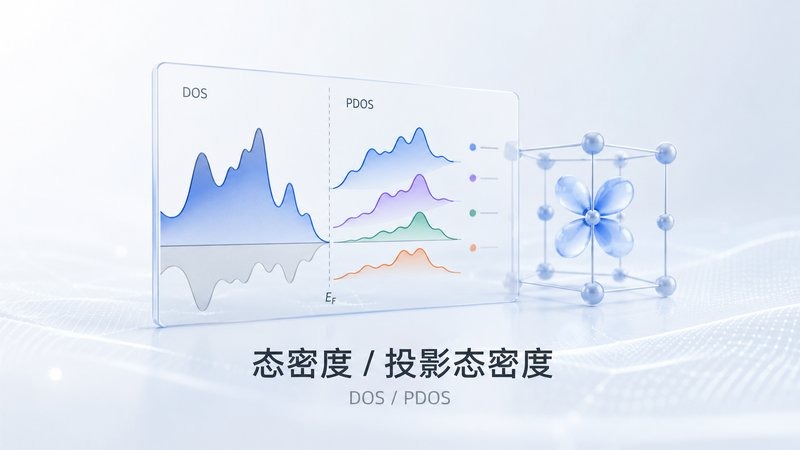 DOS / PDOS — 总态密度与轨道投影分析
DOS / PDOS — 总态密度与轨道投影分析 异质结界面 — 半导体界面电荷分离
异质结界面 — 半导体界面电荷分离晶体、表面、二维材料的第一性原理计算。覆盖能带结构、态密度、电荷密度差、COHP 成键分析、功函数等性质预测。支持最多 1000 原子周期性体系。
- 体相晶体结构弛豫与电子结构
- 表面 slab 模型构建与表面能
- 能带结构、PDOS 投影态密度
- COHP/COOP 化学键分析
- 吸附能、功函数、Bader 电荷
分子动力学与对接
:蛋白-配体复合物、膜蛋白、溶液相大分子体系(数万至数十万原子),不涉及化学反应与电子结构计算。
:经典力场分子动力学(MD)、增强采样(metadynamics、伞形采样)、MMPBSA/MMGBSA 结合自由能、分子对接。
:蛋白-配体复合物 MD 模拟、结合自由能计算、刚性/柔性/共价对接、RMSD/RMSF/氢键/距离/角度等多维度轨迹分析。
分子动力学在原子级分辨率下揭示生物大分子的动态行为,从飞秒到微秒尺度观察蛋白构象变化、配体结合/解离路径与自由能景观。配合增强采样技术可突破常规 MD 的时间尺度限制,捕获稀有事件。对接模块覆盖从单一蛋白-配体对接到大规模虚拟筛选的完整流程。
 蛋白结构 — ribbon 图展示 α 螺旋与 β 折叠
蛋白结构 — ribbon 图展示 α 螺旋与 β 折叠 分子混溶 — 分子扩散与均相混合过程
分子混溶 — 分子扩散与均相混合过程 分子聚集 — 单体→寡聚→大聚集体的自组装
分子聚集 — 单体→寡聚→大聚集体的自组装 相界面模拟 — 两相边界分子吸附与界面行为
相界面模拟 — 两相边界分子吸附与界面行为 蛋白相互作用 — 双蛋白结合界面与识别
蛋白相互作用 — 双蛋白结合界面与识别 蛋白-配体复合物 — 小分子嵌入结合口袋
蛋白-配体复合物 — 小分子嵌入结合口袋 膜蛋白体系 — 跨膜蛋白嵌入磷脂双分子层
膜蛋白体系 — 跨膜蛋白嵌入磷脂双分子层 自由能景观 — 构象空间中的能量极小值与跃迁路径
自由能景观 — 构象空间中的能量极小值与跃迁路径从常规 MD 到增强采样(metadynamics、伞形采样),覆盖溶液体系和膜蛋白环境。同时涵盖分子对接——虚拟筛选、共价对接、结合模式预测。
- 蛋白-配体 / 蛋白-蛋白复合物 MD
- MMPBSA / MMGBSA 结合自由能
- 增强采样探索自由能景观
- 刚性 / 柔性 / 共价对接
- RMSD/RMSF/氢键/距离/角度分析
机器学习 / 深度学习
:分子性质预测、材料性能回归、构效关系建模、高维数据降维与可视化。
:传统机器学习(随机森林、梯度提升树)、深度学习(图神经网络 GNN、Transformer、CNN)、模型训练与调优、数据分析与图像识别。
:分子性质预测(溶解度、毒性、活性)、材料性能预测(带隙、形成能、力学性质)、谱图识别与图像数据分析、分子指纹与描述符特征工程、模型可解释性分析(SHAP、注意力权重)。
计算化学与数据科学的交叉正在改变传统研究范式。我们专注于化学数据的深度学习建模、谱图与显微图像识别、模型训练与部署,从数据预处理、特征工程到模型评估,提供完整的 AI 辅助计算化学解决方案。
 线性回归 — 最小二乘法拟合自变量与因变量的线性关系
线性回归 — 最小二乘法拟合自变量与因变量的线性关系 K近邻 — 基于距离度量的最近邻样本投票分类
K近邻 — 基于距离度量的最近邻样本投票分类 支持向量机 — 最大间隔超平面与支持向量分类
支持向量机 — 最大间隔超平面与支持向量分类 决策树 — 层级分叉的树形分类与回归模型
决策树 — 层级分叉的树形分类与回归模型 随机森林 — 多棵决策树的集成学习与投票预测
随机森林 — 多棵决策树的集成学习与投票预测 主成分分析 — 高维数据降维与方差最大化投影
主成分分析 — 高维数据降维与方差最大化投影 卷积神经网络 — 卷积-池化层级特征提取与图像识别
卷积神经网络 — 卷积-池化层级特征提取与图像识别 图神经网络 — 图结构上的消息传递与节点表示学习
图神经网络 — 图结构上的消息传递与节点表示学习从传统 ML(随机森林、梯度提升树)到深度学习(GNN、Transformer),应用于分子性质预测、材料性能回归与分类、特征重要性分析。
- 分子性质预测(溶解度、毒性、活性)
- 材料性能预测(带隙、形成能、力学性质)
- 分子指纹与描述符特征工程
- 图神经网络 / 分子图表示学习
- 模型可解释性分析(SHAP、注意力权重)
覆盖主流计算化学工具链,
每种任务选最优工具。
以下均为免费/开源软件,团队熟练使用,根据你的科学问题选择最合适的方法和程序。
四个步骤,从结构到报告。
只接受有把握完成的任务,以最快速度、最高质量交付。
提交结构与需求
上传你的分子结构文件(PDB / SDF / XYZ / CIF),用中文描述你想回答的科学问题。
⏱ 5 分钟方案确认 & 委托
我们在 24 小时内评估计算可行性,给出推荐方法和明确的委托方案。
⏱ < 24 小时计算执行 & 实时追踪
计算执行期间保持实时沟通,关键节点同步预览确认。
⏱ 1–5 天数据交付 & 解读 + 售后
收到的不只是原始输出——包含发表级图表、SI 方法文档和中文科学解读。审稿阶段帮你专业回复。
⏱ 交付即用公式化计价,没有黑箱。
所有服务按 原子数 × 方法级别 × 加成系数 公式核算。具体标准详见委托服务手册。
你可能想问的。
更多详细内容可见委托服务手册或咨询客服。

